Are you looking for an essay on ‘Cell Cycle’? Find paragraphs, long and short essays on ‘Cell Cycle’ especially written for school and college students.
Essay on Cell Cycle
Essay Contents:
- Essay on the Introduction to Cell Cycle
- Essay on the Regulation of Cell Cycle
- Essay on the Mechanism of Cyclin -Cdk Interaction
- Essay on the Cell Cycle Checkpoints
- Essay on the Cell Cycle and Cancer
- Essay on the Apoptosis in Cells
1. Essay on the Introduction to Cell Cycle:
Growth and development is one of the main characteristics of life. It is the process of an individual organism growing organically; a purely biological unfolding of events involved in an organism changing gradually from a simple to a more complex level, while death is the permanent termination of all vital functions or life processes in an organism or cell. In general in growing condition a higher rate of anabolism is maintained than catabolism. A growing organism increases in size in all of its parts, rather than simply accumulating matter. Growth of an organism is characterized by an increase in the size of its body. In case of bacteria and other single celled organisms growth is meant by increase in number of individual cells. In biological systems growth is controlled by cell cycle systems involving mitosis and meiosis.
Cell cycle is an essential means through which a living cell divides, proliferates and propagates. In unicellular species, such as bacteria and yeasts, each cell division produces an additional organism. In multicellular species, multiple continuous cell divisions are needed to constitute living new individual. An adult human requires many millions of new cells each second in order to maintain the healthy state of the body. The duration of the cell cycle may 344 Cell and Molecular Biology
vary, however certain requirements are universal..First and foremost, to produce a pair of genetically identical daughter cells, the DNA must be replicated, and the replicated chromosomes must be segregated into two separate cells. The duration of the cell cycle varies greatly from one cell type to another. Fly embryos have the shortest known cell cycles, each lasting as little as 8 minutes, while the cell cycle of a mammalian liver cell can last longer than a year.
In 1858 the pathologist Rudolph Virchow coined the cell doctrine which states that “When a cell arises, there must have been a previous cell, just as animals can only arise from animals and plants from plants.” This doctrine is founded on the understanding that whether one is examining a single-celled organism or an animal as complex as man, the product is a result of repeated rounds of cell growth and division.
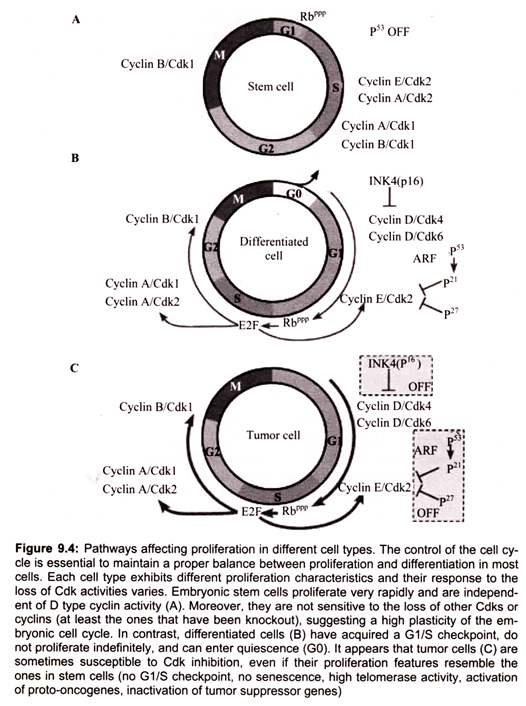
Most eukaryotic cells will proceed through an ordered series of events in which the cell duplicates its contents and then divides into two cells (Fig. 9.1). This cycle of duplication and division is called the cell cycle. In order to maintain the fidelity of the developing organism, this process of cell division in multicellular organisms must be highly ordered and tightly regulated. The loss of control will lead to abnormal development and is the cause of cancer.

The eukaryotic cell cycle is composed of five steps, or phases:
a. G0 Phase:
It is also known as quiescent stage or the resting phase. Often a cell leaves the cell cycle, temporarily or permanently at G1 stage and enters G0 phase. In general G0 cells do not reenter the cell cycle however instead continues to carry out their function in the organism until they die. For cells like lymphocyte, G0 can be followed by reentry into the cell cycle.
Most of the lymphocytes in human blood are in G0 phase. After stimulation by an antigen, they reenter the cell cycle (at G1) and proceed on to expand through new rounds of alternating S phases and mitosis. The cancerous cells never enter G0 phase and are destined to repeat the cell cycle indefinitely.
b. G1 Phase:
The first gap in the normal cell cycle is called G1 and is the period when the necessary proteins for DNA replication are synthesized. This is the first sub phase of interphase that depends in between the end of the M phase until the S phase. However, this phase of the cell cycle is not only characterized by synthesis of replication machinery. During this period the cell must monitor both the internal and external environments to ensure that all the preparations for DNA synthesis have been completed and that overall conditions for cell division are favorable.
During this phase the biosynthetic activities of the cell, which were considerably slowed down during M phase, resume activity at a high rate. During this phase various enzymes that are required in S phase, mainly those needed for DNA replication are synthesized. Here, cells increase in size and G1 associated checkpoint control mechanism and govern conditions which are appropriate favorable for DNA synthesis.
c. S Phase:
S phase is referred as synthesis phase where specifically DNA synthesis, chromosome duplication and histone proteins production takes place. Thus, the amount of DNA in the cell becomes quantitatively doubled. However, the rates of RNA transcription and protein synthesis are very low during this phase. This phase of the cell cycle is the longest taking 10-12 hours of a typical 24 hr eukaryotic cell cycle.
d. G2 Phase:
During the second gap phase of the cell cycle the cell undertakes the synthesis of the proteins required to assemble the machinery required for separation of the duplicated chromosomes (the process called mitosis) and ultimately division of the parental cell into two daughter cells (the process termed cytokinesis). Maximum protein synthesis does take place during this phase, mainly involving the production of microtubules, which are required during the process of mitosis.
Like the G1 phase, the G2 phase is also a stage when the internal and external environments are monitored to ensure that faithful replication of the DNA has occurred and that conditions are favorable for cytokinesis. The cell will continues to grow and the G2 checkpoint control mechanism ensures that cellular conditions are appropriate to enter the M (mitosis) phase and divide. Inhibition of protein synthesis during G2 phase moreover prevents the cell from undergoing mitosis.
e. M Phase:
A brief period of M phase is devoid of nuclear division, the Karyokinesis and cytoplasmic division known as cytokinesis. During M-phase there is an ordered series of events that leads to the alignment and separation of the duplicated chromosomes (called sister chromatids). This process is divided into distinct steps that were originally identified and characterized through light microscopic observations of dividing cells. The steps of mitosis are termed prophase, prometaphase, metaphase, anaphase and telophase. Although cytokinesis is the process by which the parental cell is physically separated into two new daughter cells, it actually begins during anaphase. The processes that occur during M-phase require much less time than those of S-phase, generally lasting only 1-2 hrs.
2. Essay on the Regulation of Cell Cycle:
Cell cycle research has primarily been performed on mutant strains of the fission yeast (Schizosaccharomyces pombe) and the budding yeast (Saccharomyces cerevisae) that have genetic lesions in some phase of the cell cycle. The cell division cycle (cdc) mutant strains have been quite useful in elucidating important steps. The cell cycle in yeast has two points where it is committed to proceed to the next stage in the cycle. The first point called start occurs near the end of the G1, and the cell becomes committed to DNA synthesis in the S phase of the cycle. The second commitment point is at the beginning of the M phase when the cell becomes committed to chromosomal condensation and the subsequent mitotic steps.
Cells like the skin which are constantly shed must renew themselves through the cell cycle on a regular basis. The cell cycle is also used during fetal development to allow a single fertilized egg to develop into an entire organism. Every process of the cell cycle is regulated by proteins which tell the cell what to do. These proteins are also used during interphase to confirm that conditions are appropriate for cell division. Sometimes the information is not copied exactly during interphase, and errors in a cell’s genome are created.
These errors can become harmful for the host organism, as in the case of an error which causes a cell to replicate and divide repeatedly, with no checks, forming a tumor as a cluster of cells grows out of control. Proper cell division requires a precisely ordered sequence of biochemical events that assures that every daughter cell contains a full complement of the molecules required for life. Protein kinase and protein phosphorylation are important to the timing mechanism that determines and controls entry into cell division and also ensures orderly passage through these events.
Regulation of the cell cycle involves processes crucial to the survival of a cell, including the detection and repair of genetic damage as well as the prevention of uncontrolled cell division. Regulation farther involves the role of cyclin dependent kinases (Cdks) and cyclins. By phosphorylating specific proteins at precise time intervals, these protein kinases organize and maintain the metabolic activities of the cell to produce proper cell division. The kinases are heterodimers with a regulatory subunit, cyclin, and a catalytic subunit, cyclin dependent protein kinases (CDK). In the absence of cyclin, the catalytic subunits virtually remain inactive.
When cyclin binds, the catalytic site opens up, a residue essential to catalysis which becomes virtually accessible, and the activity of catalytic subunit amplifies 10,000 folds. Animal cells have at least ten different cyclins (A, B and so forth) and at least eight different CDK (CDK1 through CDK8), which act in various combinations at specific points in the cell cycle. Plants also use a family, of CDKs to regulate their cell division. The details of cell cycle regulation, such as the number of different cyclins and kinases and the combination in which they act, differ from species to species, but the basic mechanism has been conserved in the evolution of all eukaryotic cells.
3. Essay on the Mechanism of Cyclin -Cdk Interaction:
Non-dividing (quiescent) cells (Go) enter the cell division cycle at G1, the period when the cell grows and prepares for replication. Progression of the cell cycle requires that cell pass a restriction point in Gl. Cells that do not pass through this restriction point re-enter Go. Cell cycles typically involve three additional phases, S, G2 and M. During S phase, DNA is synthesized and the centrosome is duplicated. During the M phase the cell divides (mitosis). G2 which follows the S phase is the period when the cell prepares for mitosis.
Members of the cyclin family of proteins are key regulators of the cell cycle. Cyclins bind and activate members of the cyclin-dependent kinase (Cdk) family to affect cell cycle progression. Cell cycle progression is controlled by the relative levels of individual cyclin family members. Progression through the G1-S-G2-M cycle follows successive oscillations in the levels of cyclins, D, E, A and B. Cyclins are grouped into classes that relate to the phase of the cell cycle they regulate. Cyclin D family members are G1 phase cyclins that regulate the entry of cells into G1 from Go. Cyclin D is up regulated by growth factor and external signals through the Ras GTPase signaling pathway. Cyclin D couples with Cdk4 and Cdk6.
The cyclin-D-dependent kinases enforce commitment to enter S-phase. Cyclin D- Cdk4 hypophosphorylates retinoblastoma protein (pRB) and facilitates the expression of cyclin E. Cyclin E and Cyclin A are able to bind Cdk2 and promote the cell cycle progression through G1/S transition. Cyclin E-Cdk2 and Cyclin A-Cdk2 hyperphosphorylate and inactivate pRb. The inactivation of pRb leads to activation of E2F transcription factors. Cyclin E stimulates replication complex assembly through interaction with Cdc6.
Cyclin A activates DNA synthesis by the replication complex already assembled and inhibits assembly of new replication complex. Cyclin E reinitiates the replication complex that is blocked by cyclin A. Cyclins B1 and B2 are M-phase cyclins. Cyclin B1 and cyclin B2 and their catalytic partner, Cdkl (cdc2, p34 kinase), are components of the M phase/ maturation promoting (MPF) factor that regulates processes that lead to assembly of the mitotic spindle and sister-chromatid pair alignment on the spindle.
The G1-cyclins bind to their Cdks and signal the cell to prepare the chromosomes for replication. A rising level of S-phase promoting factor (SPF) which includes cyclin A, that binds to Cdk2 enters the nucleus and prepares the cell to duplicate its DNA and centrosomes. Some cells shorten their cell cycle allowing repeated S phases without completing mitosis; this is called endoreplication for high copy number of the DNA.
Essay # 4. Essay on the Cell Cycle Checkpoints:
Cell cycle checkpoints are regulatory pathways that govern the order and timing of cell cycle transitions to ensure completion of one cellular event prior to commencement of another. The key regulators of the checkpoint pathways in the mammalian DNA damage response are the ATM (ataxia telangiectasia, mutated) and ATR (ATM and Rad3-related) protein kinases. Both of these proteins belong to a structurally unique family of serine-threonine kinases characterized by a C-terminal catalytic motif containing a phosphatidylinositol 3-kinase domain.
Although ATM and ATR appear to phosphorylate many of the same cellular substrates, they generally respond to distinct types of DNA damage. ATM is the primary mediator of the response to DNA double strand breaks (DSBs) that can arise by exposure to ionizing radiation (IR). ATR, on the other hand, plays only a back-up role in the DSB response, but directs the principle response to UV damage and stalls in DNA replication.
(i) G1 Checkpoint:
The G1 cell cycle checkpoint prevents damaged DNA from being replicated and is the best understood checkpoint in mammalian cells. Central to this checkpoint is the accumulation and activation of the p53 protein; two properties carefully controlled by the ATM and ATR kinases. In normally growing cells, p53 levels are low due to interaction with MDM2, which targets p53 for nuclear export and proteosome-mediated degradation in the cytoplasm. Following IR damage, ATM activates downstream kinase Chk2 (by phosphorylation at position T68), which in turn phosphorylates residue S20 of p53. The S20 phosphorylation of p53 blocks p53/MDM2 interaction, resulting in p53 accumulation.
ATM exerts a second control measure on p53 stability by directly phosphorylating the p53 negative regulator, MDM2, on S395. This modification allows MDM2/p53 interaction, but prevents p53 nuclear export to the cytoplasm where degradation would normally occur. The role of ATR in p53 S20 phosphorylation (and subsequent stabilization) is less well established, but implied through in vitro evidence demonstrating S20 phosphorylation by the ATR-dependent kinase, Chkl.
While phosphorylation of S20 is important to p53 stability, it is the phosphorylation of S15 that appears crucial in enhancing p53 transcriptional transactivation activity. The S15 residue of p53 can be phosphorylated directly by ATM or ATR in response to IR (ATM and ATR), UV irradiation (ATR) and stalls of DNA replication forks (ATR). Activated p53 then up-regulates a number of target genes, several of which are also involved in the DNA damage response (MDM2, GADD45a, and p21/Cip). The accumulation of p21, a cyclin- dependent kinase inhibitor, suppresses Cyclin E/Cdk2 kinase activity thereby resulting in G1 arrest (Fig. 9.5).

Induced or spontaneous DNA lesions are common events in the life of the cell. The ability of the cell to maintain homeostasis and protect itself from neoplastic transformation depends upon complex surveillance mechanisms and activation of repair pathways to preserve chromosomal integrity. The DNA damage checkpoint is a cardinal process. Genetic defects that perturb DNA repair mechanisms almost always cause severe diseases, including ataxia-telangiectasia and related syndromes, characterized by degeneration of the nervous and immune systems, sensitivity to ionizing radiation and DNA-damaging agents, and predisposition to cancer.
The serine-threonine protein kinases ataxia telangiectasia mutated (ATM; also known as serine protein kinase ATM) and ataxia telangiectasia and RAD3-related protein (ATR; also known as serine-threonine protein kinase ATR) are DNA damage sensor proteins that can induce cell cycle arrest, DNA damage repair or apoptosis, depending on the extent of the DNA lesions, whereas ATM responds primarily to DNA double-strand breaks, which are generally caused by ionizing radiation and radiomimetic drugs, ATR also responds to damage caused by ultraviolet light and stalled replication forks.
The ATM-ATR cascade is activated within minutes of a DNA damage alarm (Fig. 9.5). Both ATM and ATR can phosphorylate and activate the transcription factor p53, either directly or by means of prior activation of checkpoint kinase 2 (CHK2). Among the genes induced by p53 is the cyclin-dependent kinase 2 (CDK2) inhibitor p21 (also known as CDKN1A and CIP1), the activity of which prevents damaged cells from entering the DNA synthesis (S) phase.
Also, damaged cells that have already passed the transition from the first gap (G1) phase to S phase can be halted through the activation of another ATM-ATR effector, CHK1, which phosphorylates the dual-specificity phosphatase CDC25C, providing a signal that induces its sequestration in the cytoplasm. Because CDC25C is responsible for removing two inhibitory phosphates from CDK1, its inactivation prevents the cell from entry into the mitosis (M) phase. Cell cycle arrest in G1, S or G2 phase is maintained until DNA integrity is restored. If lesions are irreparable, programmed cell death is induced by the ATM -ATR signalling pathway. The ATM-CHK2 pathway predominantly regulates the G1 checkpoint, whereas the ATR-CHK1 pathway predominantly regulates the S and G2 checkpoints, although there is crosstalk between these pathways.
In most human cancers, however, the function of the DNA damage checkpoint in G1 is impaired owing to mutations in p53 or the gene encoding the retinoblastoma protein (RBI). Treatment of these tumour cells with DNA-damaging agents, such as ionizing radiation and DNA-targeting drugs, results in S or G2 checkpoint-mediated arrest. Nonetheless, some of these cells might use this remaining checkpoint to protect themselves from radiation or cytotoxic agents.
These cancer-favouring circumstances may be tackled by the combination of DNA-damaging drugs or ionizing radiation with inhibitors of the S or G2 checkpoints, or ‘S or G2 checkpoint abrogators’. Such a combination should force cancer cells carrying DNA lesions into mitosis, a condition which prompts mitotic catastrophe and associated cell death. Abrogation of the DNA damage checkpoint in S or G2 is an attractive strategy for selectively targeting G1 checkpoint-defective cancer cells and is currently being explored in clinical trials.
p53:
It is a tumor suppressor gene. The p53 protein senses DNA damage and halts progression of the cell cycle in G1 by blocking the activity of cdk2. The p53 protein also plays a key role in apoptosis by forcing unfunctional or bad cells to commit suicide. p53 is a protein that functions to block the cell cycle if the DNA is damaged. If the damage is severe this protein can cause apoptosis (cell death).
1. p53 levels are increased in damaged cells. This allows time to repair DNA by blocking the cell cycle.
2. A p53 mutation is the most frequent mutation leading to cancer. An extreme case of this is Li Fraumeni syndrome, where a genetic defect in p53 leads to a high frequency of cancer in affected individuals.
One major function of the p53 protein, which is active as a homotetrameric transcription factor, is to serve as a component of the checkpoint that controls whether cells enter as well as progress through S-phase. The action of p53 is induced in response to DNA damage. Under normal circumstances p53 levels remain very low due to its interaction with a member of the ubiquitin ligase family called MDM2. MDM2 is so named since it was isolated as an amplified gene in the tumorigenid mouse cell line 3T3DM.
In response to DNA damage, e.g. as a result of UV-irradiation or y-irradiation, cells activate several kinases including checkpoint kinase 2 (ChK2) and ataxia telangiectasia mutated (ATM). One target of these kinases is p53. ATM also phosphorylates MDM2. When p53 is phosphorylated it is released from MDM2 and can carry out its transcriptional activation functions. One target of p53 is the cyclin inhibitor p21Cip1 gene. Activation of p21cip1 leads to increased inhibition of the cyclin D1-CDK4 and cyclin E-CDK2 complexes thereby halting progression through the cell cycle either prior to S-phase entry or during S-phase.
As a consequence of p53-induced synthesis of p21 expression, there is a convergence between the roles of p53 and pRB in regulation of cyclin-CDK complexes. In either case the aim is to allow the cell to repair its damaged DNA prior to replication or mitosis.
(ii) S-Phase Checkpoint:
The S-phase checkpoint monitors cell cycle progression and decreases the rate of DNA synthesis following DNA damage. Although, this pathway is the least understood of the mammalian checkpoints, recent studies on IR-induced S-phase checkpoint activation are beginning to provide important insight. Cells from cancer-prone individuals affected with ataxia telangiectasia (AT) or Nijmegen breakage syndrome (NBS) fail to slow their rate of DNA replication following IR exposure; a phenomenon known as radio-resistant DNA synthesis (RDS). This finding implicates the associated gene products (ATM and NBS1, respectively) in the S-phase checkpoint pathway.
Experimental evidence indicates that IR damage activates the S-phase checkpoint via at least 2 parallel branches, both of which are regulated by ATM. In the first branch, IR damage induces the phosphorylation of the Chk2 kinase (at T68) by ATM. Chk2, once activated, targets the Cdc25A phosphatase for ubiquitin- dependent degradation by phosphorylating it on SI23. The resultant destabilization of Cdc25A prevents it from performing its normal function of removing inhibitory phosphorylations (T14 and Y15) from Cdk2. The Cdk2/Cyclin E and Cdk2/Cyclin A complexes remain inactive thus preventing completion of DNA synthesis.
The second branch of the IR-induced S-phase checkpoint pathway is independent of Cdc25A, but requires the activities of both ATM and NBS1. Upon IR damage, ATM phosphorylates a number of downstream substrates including NBS1 (at multiple sites including S343), the product of the breast cancer susceptibility gene 1 (BRCA1; at multiple sites including SI387), and SMC1 (structural maintenance of chromosome protein 1; at S957 and S966). Loss of any of these proteins or mutation of the indicated phosphorylation sites results in attenuated S-phase checkpoint activation.
The involvement of ATR in the S-phase checkpoint also remains relatively obscure. ATR has been shown to initiate a slow IR- induced S-phase checkpoint response by phosphorylating its effector kinase, Chk1 (on S317 and S345), which in turn phosphorylates Cdc25A targeting it for degradation. In addition, SMC1 undergoes S957 and S966 phosphorylation upon UV irradiation or hydroxyurea treatment in an ATM-independent manner.
(iii) G2 Checkpoint:
The G2 cell cycle checkpoint is an important control measure that allows suspension of the cell cycle prior to chromosome segregation. Entry into mitosis is controlled by the activity of the cyclin dependent kinase Cdc2. Maintenance of the inhibitory phosphorylations on Cdc2 (on T14 andY15) is essential for G2 checkpoint activation. ATM and ATR indirectly modulate the phosphorylation status of these sites in response to DNA damage. Unlike other checkpoints, the response to IR is mediated primarily by ATR with ATM playing a back-up role; the response to UV damage and replication blocks is controlled by ATR.
It should be noted that the stage of the cell cycle when the DNA damage occurs may influence whether the response is mediated through ATR or ATM. In any case, upon DNA damage, the downstream kinases Chk1 and Chk2 (activated by ATR- and ATM-dependent phosphorylation, respectively) phosphorylate the dual specificity phosphatase Cdc25C on position S216. Phosphorylation of this residue creates a binding site for the 14-3-3 proteins. The 14-3-3/ Cdc25C protein complexes are sequestered in the cytoplasm, thereby preventing Cdc25C from activating Cdc2 through removal of the T14 and Y15 inhibitory phosphorylations. This results in the maintenance of the Cdc2/Cyclin B1 complex in its inactive state and blockage of entry into mitosis.
DNA damage checkpoints sense and detect DNA damage both before a cell enters S phase (a G1 checkpoint) as well as after S phase (a G2 checkpoint). Damage to DNA before the cell enters S phase inhibits the action of cdk2 thus stopping the progression of the cell cycle until the damage is repaired with the help of BRCA2. The BRCA2 gene (Breast Cancer Type 2 susceptibility protein) belongs to a class of genes known as tumor suppressor genes. Like many other tumor suppressors, the protein produced from the BRCA2 gene helps prevent cells from growing and dividing too rapidly or in an uncontrolled way. The BRCA2 gene provides instructions for making a protein that is directly involved in the repair of damaged DNA.
In the nucleus of many types of normal cells, the BRCA2 protein interacts with several other proteins, including the proteins produced from the RAD51 and PALB2 genes, to prevent breaks in DNA. These breaks can be caused by natural and medical radiation or other environmental exposures, and also occur when chromosomes exchange genetic material in preparation for cell division. By helping repair DNA, BRCA2 plays a role in maintaining the stability of a cell’s genetic information. If the damage is severe, while cannot be repaired, the cell undergoes destruction following apoptosis. Damage to DNA after S phase (the G2 checkpoint), inhibits the action of cdk1 thus prevents the cell from proceeding from G2 to mitosis.
In addition to intrinsic controls exerted by CDKs and checkpoints, many external controls affect cell division. Both normal and abnormal cell cycles can be triggered by such extrinsic controls. For example, the hormone estrogen affects the development of a wide variety of cell types in women. Estrogen exerts its effects on a receptive cell by binding to a specific receptor protein on the cell’s nuclear membrane. By binding to an estrogen receptor, estrogen initiates a cascade of biochemical reactions that lead to changes in the cell-cycle program. Normally, estrogen moves cells out of a resting stage into an active cell cycle.
In a different context, however, even normal levels of estrogen encourage the growth of some forms of breast cancer. In these cases, estrogen increases the speed with which the cancerous cells complete their cell cycles, leading to more rapid growth of the tumor. The most effective current drug therapies for such breast cancers block the estrogen receptors estrogen binding ability, making cells unresponsive to estrogen’s proliferation signal. Thus, while estrogen itself does not cause breast cancer, it plays an important role in stimulating the growth of some cancers once they initiate by other mechanisms, such as by an unregulated CDK or a defect in a cell-cycle checkpoint.
Certain cyclins are present only during certain stages of the cell cycle. There are several cyclin-dependent kinases and several cyclins which control the G1 checkpoint (G1 Cdk’s and G1 cyclins) and the G2 checkpoint (mitotic Cdk’s and mitotic cyclins). Once bound to the cyclin, the mitotic cyclin-dependent kinase complex (or MPF) phosphorylates proteins involved in the early stages of mitosis. The active MPF stimulates the breakdown of the nuclear envelope, chromosome condensation, mitotic spindle formation and degradation of key proteins.
The mitotic Cdk-cyclin complex (MPF) also controls the spindle assembly checkpoint by activating the anaphase promoting complex. The DNA damage checkpoint is regulated by p53, “the Guardian of the Genome”. The G1 Cdk-cyclin complex controls the G1 checkpoint by the phosphorylation of a number of proteins. One main target is the retinoblastoma (Rb) protein. Phosphorylation of Rb prevents the binding and inactivation of the transcription factor E2F. When E2F is active it allows the transcription of a number of gene products that are essential to trigger S phase. During M phase, Rb is dephosphorylated and E2F is inhibited. In summary, the cell cycle is regulated by the control of checkpoints by cyclin-Cdk complexes (Fig. 9.6).

5. Essay on the Cell Cycle and Cancer:
Animals are multicellular organisms that require the normal function of all the organs of the body. These organs are developed from different tissues and each of the tissues is products of cell division. For the body to function normally, the organs and tissues must communicate to control the development of the cells and tissues. Otherwise, uncontrolled cell growth in one part of the body could infringe on the development of other cells or tissues.
Then the normal functions of the individual would be seriously impaired. Oncogenes have been shown many times to be associated with cancer and uncontrolled cellular growth. This growth can lead to two types of tumors. Two types of tumors exist. Malignant tumors can induce secondary tumors by the release of cells that can lodge and begin growing in another location of the body. Benign tumors are cells that remain in the initial location.
The cell division process is dependent on a tightly controlled sequence of events. These events are dependent on the proper levels of transcription and translation of certain genes. When this process does not occur properly, unregulated cell growth may be the end result. Of the 30,000 or so genes that are currently thought to exist in the human genome, there is a small subset that seems to be particularly important in the prevention, development, and progression of cancer. These genes have been found to be either malfunctioning or nonfunctioning in many different kinds of cancer.
The genes that have been identified to date have been categorized into two broad categories, depending on their normal functions in the cell such as the genes, whose protein products stimulate or enhance the division and viability of cells. This first category also includes genes that contribute to tumor growth by inhibiting cell death.
The second one are the genes whose protein products can directly or indirectly prevent cell division or lead to cell death. The normal versions of genes in the first group are called proto-oncogenes. The mutated or otherwise damaged versions of these genes are called oncogenes. The genes in the second group are called tumor suppressors.
The dis-regulation of the cell cycle components may lead to tumor formation. Some genes such as the cell cycle inhibitor p53 (tumor suppressor gene whose protein senses DNA damage and can halt progression of the cell cycle in G1 phase), when mutates, may cause the cell to multiply uncontrollably, forming a tumor. Although, the duration of cell cycle in tumor cells is equal to that of normal cell cycle, the proportion of cells that are in active cell division versus quiescent cells in Go phase in tumors, is much higher than that in normal tissue because the cancerous cells rarely enter the resting phase. Thus there is a net increase in cell number and mass as the number of cells that die by apoptosis or senescence remains the same.
The proteins suppress the ability of cancer to develop are known as tumor suppressors because cancer ensues as a result of a loss of their normal function. It would seem obvious; therefore, that one import function of tumor suppressors would be control of the progression of a cell through a round of the cell cycle. If cells are able to synthesize damaged DNA before it is repaired or to divide when the DNA is damaged then the resulting daughter cells can pass on the resultant DNA damage to their progeny.
The result can be catastrophic resulting in cancer. For this reason, the two most important check points in the eukaryotic cell cycle are the G1-S transition and the entry into mitosis. The former prevents DNA replication prior to repair of damaged DNA and the latter prevents damage that may have occurred to the DNA during replication to propagate into daughter cells during mitosis.
Following the isolation and characterization of two tumor suppressor genes in particular it was found that they function to control the ability of cells to progress through these two important checkpoints. The protein encoded by the retinoblastoma susceptibility gene (pRB) and the p53 protein are both tumor suppressors. The function of pRB is to act as a brake preventing cells from exiting G1 and that of p53 is to inhibit progression from S-phase to M-phase.
6. Essay on the Apoptosis in Cells:
Apoptosis is the term given when programmed cell death (PCD) occurs in multicellular organisms. Apoptosis is one of the main types of programmed cell death which involves a series of biochemical events leading to specific cell morphology characteristics and ultimately death of cells. The word apoptosis has ancient Greek origins, referring to the falling of leaves, or possibly “dropping of scabs” or “falling off of bones. “Characteristic cell morphology of cells undergoing apoptosis include changes to the cell membrane such as loss of membrane asymmetry and attachment, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation. Apoptosis differentiates from necrosis as the processes associated with apoptosis in disposal of cellular debris do not damage the organism in apoptosis.
Cells die in response to a variety of stimuli and during apoptosis they do so in a controlled, regulated fashion. This makes apoptosis distinct from another form of cell death called necrosis in which uncontrolled cell death leads to lysis of cells, inflammatory responses and, potentially, to serious health problems. Apoptosis, by contrast, is a process in which cells play an active role in their own death (which is why apoptosis is often referred to as cell suicide).
This process of cell death has been termed also programmed cell death or active cell death because it requires controlled gene expression, which is activated in response to a variety of external or internal stimuli or their absence. Upon receiving specific signals instructing the cells to undergo apoptosis a number of distinctive changes occur in the cell. A family of proteins known as caspases is typically activated in the early stages of apoptosis. These proteins breakdown or cleave key cellular components that are required for normal cellular function including structural proteins in the cytoskeleton and nuclear proteins such as DNA repair enzymes. The caspases can also activate other degradative enzymes such as DNases, which begin to cleave the DNA in the nucleus.
Cell death can be triggered by a variety of stimuli, including gamma irradiation, cytotoxic lymphocytes, glucocorticoids, and various cytolytic cytokines, for example, TNF- alpha. In thymocytes apoptosis can be induced by treatment with glucocorticoids or irradiation. Many growth factors and cytokines act as cellular survival factors by preventing apoptosis. They are said to have anti-apoptotic activities. Apoptosis can also be initiated by cross- linking or engagement of one of several death receptor surface antigens. Some cell types such as neutrophils or eosinophils are constitutively programmed to undergo cell death by apoptosis.
In some cases the apoptotic stimuli comprise extrinsic signals such as the binding of death inducing ligands to cell surface receptors called death receptors. These ligands can either be soluble factors or can be expressed on the surface of cells such as cytotoxic T lymphocytes. The latter occurs when T-cells recognize damaged or virus infected cells and initiate apoptosis in order to prevent damaged cells from becoming neoplastic (cancerous) or virus-infected cells from spreading the infection. Apoptosis can also be induced by cytotoxic T-lymphocytes using the enzyme granzyme.
In other cases apoptosis can be initiated following intrinsic signals that are produced following cellular stress. Cellular stress may occur from exposure to radiation or chemicals or to viral infection. It might also be a consequence of growth factor deprivation or oxidative stress caused by free radicals. In general intrinsic signals initiate apoptosis via the involvement of the mitochondria. The relative ratios of the various bcl-2 proteins can often determine how much cellular stress is necessary to induce apoptosis.
Many physiological processes, including proper tissue development and homeostasis, require a balance between apoptosis and cell proliferation. All somatic cells proliferate via a mitotic process determined by progression through the cell cycle. Apoptosis (programmed cell death) occurs in a wide variety of physiological settings, where its role is to remove harmful, damaged or unwanted cells (Fig. 9.7). Apoptosis and cell proliferation are linked by cell-cycle regulators and apoptotic stimuli that affect both processes. As somatic cells proliferate, the cell-cycle progression is regulated by positive and negative signals. Apoptosis and mitosis share common morphological features such as cell shrinkage, chromatin condensation and membrane blebbing. Additionally, cell-cycle genes such as p53, RB and E2F have been shown to participate in both the cell cycle and in apoptosis. Thus, the balance between apoptosis and proliferation must be strictly maintained to sustain tissue homeostasis.

A damaged cell may undergo apoptosis if it is unable to repair genetic errors. There seem to be two major reasons for apoptosis. First, apoptosis is one means by which a developing organism shapes its tissues and organs. For instance, a human fetus has webbed hands and feet early on its development. Later, apoptosis removes skin cells, revealing individual fingers and toes. A fetus’s eyelids form an opening by the process of apoptosis. During metamorphosis, tadpoles lose their tails through apoptosis. In young children, apoptosis is involved in the processes that literally shape the connections between brain cells, and in mature females, apoptosis of cells in the uterus causes the uterine lining to slough off at each menstrual cycle.
Cells may also commit suicide in times of distress, for the good of the organism as a whole. For example, in the case of a viral infection, certain cells of the immune system, called cytotoxic T lymphocytes, bind to infected cells and trigger them to undergo apoptosis. Also, cells that have suffered damage to their DNA, which can make them prone to becoming cancerous, are induced to commit apoptosis.
Malfunctions of apoptosis have been implicated in many forms of human diseases such as neurodegenerative diseases, AIDS and ischemic stroke. Reportedly, apoptosis is caused by various inducers such as chemical compounds, proteins or removal of NGF. The biochemical pathways of apoptosis are complex and depend on both the cells and the inducers.