Are you looking for an essay on ‘Genetic Engineering’? Find paragraphs, long and short essays on ‘Genetic Engineering’ especially written for college and medical students.
Essay on Genetic Engineering
Essay Contents:
- Essay on the Introduction to Genetic Engineering
- Essay on the Essential Steps of Genetic Engineering
- Essay on the Molecular Tools in rDNA Technology
- Essay on the Vectors – The Cloning Vehicle
- Essay on the Methods of Gene Transfer
- Essay on the Recombinant Selection
- Essay on the Expression Systems
- Essay on the Production and Purification of rDNA Products
- Essay on Transgenic Plants
- Essay on the Edible Vaccines
- Essay on Gene Therapy
1. Essay on the Introduction to Genetic Engineering:
Genetic engineering is relatively a new discipline of science which is used highly controllable laboratory conditions to alter the heredity apparatus of a living cell (i.e., the manipulation of genes under highly controllable laboratory conditions) so that the cell can produce different chemicals, or perform completely new functions. It is perhaps the finest of biologists who have been successful in tailoring and manipulating heredity to the best application to the mankind.
The onset of this new revolutionary technology of genetic engineering took place at Massachusetts Institute of Technology (MIT), USA when H.G. Khorana, scientist of, Indian origin along with his colleagues in 1972 first reported total synthesis of an artificial gene, namely, tyrosine tRNA gene, with the potential for functioning within a living cell.
Research on genetic engineering is essentially involves in vitro joining or recombination of DNA fragments of different origins with the help of some highly specific enzymes to produce ‘recombinant-DNA’ which is subsequently introduced into appropriate hosts where it multiples (gene cloning). Thus the genetic engineering is also called “recombinant-DNA technology”. Thus a multitude of possibilities prevails to bring about new gene combinations not occurring in nature. The science of recombinant-DNA technology or genetic engineering is still its infancy. Even then, its enormous potential impact both social and commercial, are clearly evident.
At present the microorganisms dominate in the sphere of this fascinating technology. Most of the basic information regarding this novel approach has been through microorganisms. Restriction enzymes, the enzymes that cleave DNA molecules to obtain desired genes, are obtained from microorganisms and so are the ligases that are used in joining the DNA fragments. Almost all the vectors, the vehicles that carry genes (DNA fragments) from donor DNA to recipient DNA, are microbial; for example, plasmids, cosmids and viral vectors. Not only these but, there are microorganisms which are predominantly cloned with new genes to produce products of choice.
Recombinant DNA works when the protein is expressed from the recombinant gene by host cell. A significant amount of recombinant protein will not be produced by the host unless expression factors are added. Protein expression depends upon the gene being influenced by a number of signals which provide instructions for the transcription and translation of the gene by the cell.
These signals include the promoter, the ribosome binding site, and the terminator. Expression vectors, in which the foreign DNA is inserted, contain these signals. Signals are species specific. In the case of E. coli, these signals must be E. coli signals as E. coli is unlikely to follow the signals of human promoters and terminators.
The presence of introns or signals which act as terminators to a bacterial host in the gene of interest may results in premature termination, and the recombinant protein may expressed and processed improperly like folding, or even may suffer degradation. The preferential eukaryotic system for the production of recombinant proteins is yeast and filamentous fungi. Some proteins are too complex to be produced in bacterium, so eukaryotic cells must be used. Animal cells can also be used for this purpose but the main hindrances are its complex growth requirements such as solid support surface, unlike bacteria and other growth conditions.
2. Essay on the Essential Steps of Genetic Engineering:
Genetic engineering involves the isolation of required genes, their insertion into a bacterial cell (cloning organism) and allowing the gene to replicate along with the genes of bacterial cell. Each step needs ultimate precision and care for the desired results.
The various techniques used in each step can be summarized as follows (Fig. 10.9):


A. Isolation of DNA Molecule Containing the Required Gene:
The gene of interest can be obtained either form a gene library or can be a PCR product. It can also obtain from natural sources by breaking the cell and isolating the DNA and its modification by restriction enzymes. The presence of gene of interest can be easily detected by various hybridization techniques.
B. Construction of Recombinant-DNA Molecule:
The construction of recombinant-DNA molecule (hybrid DNA molecule) is accomplished by the use of two enzymes: restriction enzymes and DNA ligases. The isolated DNA molecule containing the gene of interest (required gene) is treated with a restriction enzyme which cleaves DNA molecule to yield fragments of various sizes, one of which represents the required gene, alternatively, the required gene may also be synthesized artificially with the help of gene machine or automated polynucleotide synthesizer.
Simultaneously, a cloning vector (may be generally a plasmid or a bacteriophage) is also treated with the same restriction enzymes. A gap is created in plasmid where the required gene gets inserted. The required gene (or gene of interest) is now incorporated in the gap created in the cloning vector through co-incubating. The vector and target genes are combined by enzyme DNA ligase in a resulting-DNA molecule or hybrid DNA molecule.
C. Insertion of Recombinant DNA:
The recombinant-DNA molecule is inserted into a competent host cell which is usually a bacterium. A competent host cell is one which accepts the recombinant-DNA molecule and allows it to replicate autonomously within it. The cell that accepts the recombinant-DNA molecule is called cloning organism which, finally, contains its own genes as well as the incorporated gene of interest.
D. Production of Clones:
The final step in gene cloning is to allow the cloning organisms to multiply. The cloning organism is plated out on to agar medium where it divides. When it grows and divides, it yields a colony of identical cells possessing equivalent genetic, components and therefore physiological traits, such a colony is referred to as a clone as it has originated from a single cell and consisted of all identical cells. All cells of a clone, like the cloning organism, contain the required gene or the gene of interest. However, the cells of a clone are grown in limitless quantity to amplify the required gene and, therefore, the product of the required gene.
3. Essay on the Molecular Tools in rDNA Technology:
A. Cutting and Joining DNA:
Two major categories of enzymes represent the biological tools of importance critically used in the isolation of DNA and the preparation of recombinant DNA, restriction endonucleases and DNA ligases. Restriction endonucleases recognize a specific, rather short, nucleotide sequence on a double-stranded DNA molecule, called a restriction site, and cleave the DNA at this recognition site or elsewhere, depending on the type of enzyme. DNA ligase on the contrary joins two pieces of DNA by forming phosphodiester bonds.
B. Major Classes of Restriction Endonucleases- DNA Cutting Enzyme:
Restriction enzymes protect bacteria from infections by viruses, and it is generally accepted that this is their role in nature. They function as microbial immune systems. When a strain of E. coli lacking a restriction enzyme infected with a virus, most virus particles can a successful infection. Restriction enzymes were discovered 40 years ago during investigations of the phenomenon of host-specific restriction and modification of bacterial viruses. Among the first restriction enzymes to be purified were EcoRl and EcoRII from Escherichia coli, and Hindll and Hindlll from Haemophilus influenzae. These enzymes were found to cleave DNA at specific sites, generating discrete, gene-size fragments that could be rejoined in the laboratory.
Restriction enzymes are generally classified into four types on the basis of their subunit composition, cleavage position, sequence specificity and cofactor requirements (Table 10.1).

Type I enzymes are complex, multisubunit, combination restriction-and-modification enzymes that cut DNA at random far from their recognition sequences. Originally, thought to be rare, we now know from the analysis of sequenced genomes that they are common. Type I enzymes are of considerable biochemical interest, but they have little practical value since they do not produce discrete restriction fragments or distinct gel-banding patterns.
Type II enzymes cut DNA at defined positions close to or within their recognition sequences. They produce discrete restriction fragments and accordingly distinct gel banding patterns. Thus they are used in the laboratory for DNA analysis and gene cloning. Rather than forming a single family of related proteins, Type II enzymes are a collection of unrelated proteins of many different sorts. Type II enzymes frequently differ in amino acid sequence from one another, and indeed from every other known protein, that they exemplify and represent for a class of rapidly evolving proteins that are often indicative of involvement in host-parasite interactions.
Type II restriction endonuclease is composed of two identical polypeptide subunits that join together to form a homodimer. These homodimers recognize short symmetric DNA sequences of 4-8 bp. six base pair cutters are the most commonly used in molecular biology research. Usually, the sequence read in the 5→ 3direction on one strand is the same as the sequence read in the 5→ 3direction on the complementary strand. Sequences that read the same in both directions are called palindromes (from the Greek word palindromes for “run back”).
Some enzymes, such as ZTcoRl, generate a staggered cut, in which the single- stranded complementary tails are called “sticky” or cohesive ends because they can form hydrogen bond with the single-stranded complementary tails of other DNA fragments. If DNA molecules from different sources share the same palindromic recognition sites, both will contain complementary sticky ends (single-stranded tails) when digested with the same restriction endonuclease.
Other type II enzymes, such as SmaI, cut both strands of the DNA at the same position and generate blunt ends with no unpaired nucleotides when they cleave the DNA. Restriction endonucleases exhibit a much greater degree of sequence specificity in the enzymatic reaction than is exhibited in the binding of other regulatory proteins.
All structures of orthodox type II restriction endonucleases characterized by X-ray crystallography so far show a common structural core composed of four conserved β -strands and one α-helix. In the presence of the essential cofactor Mg2+, the enzyme cleaves the DNA on both strands at the same time within or in close proximity to the recognition sequence (restriction site). The enzyme cuts the DNA duplex by breaking the covalent, phosphodiester bond between the phosphate of one nucleotide and the sugar of an adjacent nucleotide, to give free 5-phosphate and 3-OH ends. Type II restriction endonucleases do not require ATP hydrolysis for their nucleolytic activity.
Type III enzymes are also large combination of restriction-and-modification enzymes. They cleave outside of their recognition sequences and require two such sequences in opposite orientations within the same DNA molecule to accomplish cleavage; they rarely give complete digests.
Type IV enzymes recognize modified, typically methylated DNA and are exemplified by the McrBC and Mrr systems of E. coli.
C. Patterns of DNA Cutting by Restriction Enzymes:
Restriction enzymes hydrolyze the backbone of DNA between deoxyribose and phosphate groups. This leaves a phosphate group on the 5′ ends and a hydroxyl on the 3′ ends of both strands. A few restriction enzymes, however cleave single stranded DNA, although usually at low efficiency. Most of the restriction enzymes used in molecular biology labs cut within their recognition sites and generate one of three different types of ends.
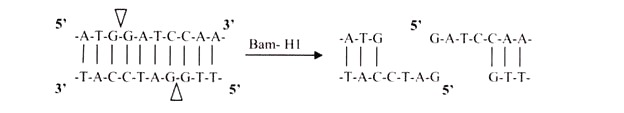
i. 5′ Overhangs:
The enzyme cuts asymmetrically within the recognition site such that a short single-stranded segment extends from the 5′ ends. BamHI cuts in this manner.
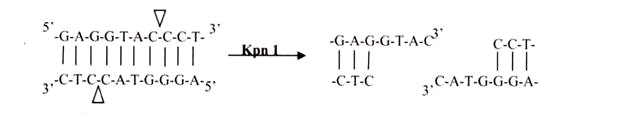
ii. 3′ Overhangs:
Again, there is asymmetrical cutting within the recognition site, but the result is a single-stranded overhang from the two 3′ ends. Kpnl cuts in this manner.
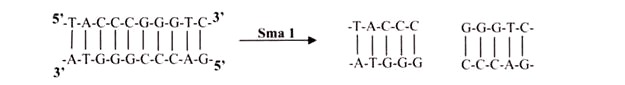
iii. Blunts:
Enzymes that cut at precisely opposite sites in the two strands of DNA generate blunt ends without overhangs. SmaI is an example of an enzyme that generates blunt ends. The 5′ or 3’ overhangs generated by enzymes that cut asymmetrically are called sticky ends or cohesive ends, because they readily stick or anneal with their partner by base pairing.
D. Restriction Endonclease Nomenclature:
Restriction endonucleases are named by a standard procedure, with particular reference to the bacteria from which they are isolated. The first latter (in italics) of the enzyme indicates the genus name, followed by the first two letters (also in italics) of the species, then comes the strain of the organism and finally a Roman numeral indicating the order of discovery. For example, Hindlll (pronounced “hindee-three”) was discovered in Haemophilus influenza (strain d).
The Hin comes from the first letter of the genus name and the first two letters of the species name; d is for the strain type; and III is for the third enzyme of that type. Smal is from Serratia marcescens and is pronounced “smah-one,” JEcoRI (pronounced “echo-r-one”) was discovered in Escherichia coli (strain R), and BamHI is from Bacillus amyloliquefaciens (strain H). Over 3000 type II restriction endonucleases have been isolated and characterized to date. Approximately 240 are available commercially for use by molecular biologists (Table 10.2).

E. DNA Ligase:
The study of DNA replication and repair processes led to the discovery of the DNA-joining enzyme called DNA ligase. DNA ligases catalyze formation of a phosphodiester bond between the 5′-phosphate of a nucleotide on one fragment of DNA and the 3′-hydroxyl of another. This joining of linear DNA fragments together with covalent bonds is called ligation. Unlike the type II restriction endonucleases, DNA ligase requires ATP as a cofactor. As it can join two pieces of DNA, DNA ligase became a key enzyme in genetic engineering. If restriction-digested fragments of DNA are placed together under appropriate conditions, the DNA fragments from two sources can anneal to form recombinant molecules by hydrogen bonding between the complementary base pairs of the sticky ends. However, the two strands are not covalently bonded by phosphodiester bonds.
DNA ligase is required to seal the gaps, covalently bonding the two strands and regenerating a circular molecule. The DNA ligase most widely used in the lab is derived from the bacteriophage T4. T4 DNA ligase will also ligate fragments with blunt ends, but the reaction is less efficient and higher concentrations of the enzyme are usually required in vitro. To increase the efficiency of the reaction, researchers often use the enzyme terminal deoxynucleotidyl transferase to modify the blunt ends by homopolymer tailing.
F. Homopolymer Tailing:
This mechanism of homopolymer tailing is joining of complementary DNA stands by annealing. For example, if a single-stranded poly(dA) tail is added to DNA fragments from one source, and a single stranded poly(dT) tail is added to DNA from another source, the complementary tails can combine together through hydrogen bonds. Recombinant DNA molecules can then be created by ligation. The homopolymer extension (by adding 10-40 residues) can be synthesized by using terminal deoxy nucleotidyltransferase (of calf thymus).
G. Linker and Adaptor:
Linker and adaptor are chemically synthesized, short, double stranded DNA possessing multiple cloning sites (also called the polylinker region) which have a number of unique target sites for restriction endonucleases. Cutting the circular plasmid vector with one of these enzymes results in a single cut, creating a linear plasmid. A foreign DNA molecule, referred to as the “insert,” cut with the same enzyme, can then is joined to the vector in ligation reaction. They can be ligated to blunt ends of any DNA molecule and cut with specific restriction enzyme to produce DNA fragments with sticky ends. Adaptors are useful to be ligated to DNA fragments with blunt ends. The DNA fragments held to linker or adaptors are finally ligated to vector DNA molecule.
Ligations of the insert to vector are not 100% productive, because the two ends of a plasmid vector can be readily ligated together, which is called self-ligation. The degree of self-ligation can be reduced by treatment of the vector with the enzyme alkaline phosphatase. Alkaline phosphatase is an enzyme which removes the terminal 5′-phosphate. When the 5′-phosphate is removed from the plasmid it cannot be re-circularized by ligase, since there is nothing with which to make a phosphodiester bond. But, if the vector is joined with a foreign insert, the 5′-phosphate is provided by the foreign DNA. Another strategy involves using two different restriction endonuclease cutting sites with non-complementary sticky ends. This inhibits self-ligation and promotes annealing of the foreign DNA in the desired orientation within the vector.

4. Essay on the Vectors – The Cloning Vehicle:
Vectors are the DNA molecules, which carry a foreign DNA fragment to be cloned. The ideal characteristics of a cloning vector are that they can independently replicate themselves as well as the foreign DNA segments they carry. They contain a number of unique restriction endonuclease cleavage sites that are present only once in the vector. They carry a selectable marker (usually in the form of antibiotic resistance genes or genes for enzymes missing in the host cell) to distinguish host cells that carry vectors from host cells that do not contain a vector and are relatively easy to recover from the host cell. The selection and choices of vector are depends on the purpose of cloning.
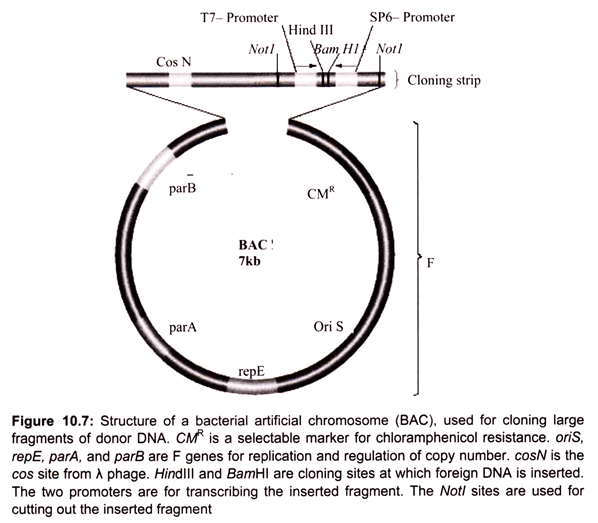
The classic cloning vectors are plasmids, phages, and cosmids, which are limited to the size insert they can accommodate, taking up to 10, 20, and 45 kb, respectively. A cosmid is a plasmid carrying a phage λ cos site, allowing it to be packaged into a phage head. Cosmids infect a host bacterium as do phages, but replicate like plasmids and the host cells are not lysed. Mammalian genes are often greater than 100 kb in size, so originally there were limitations in cloning complete gene sequences (Table 10.3). This new generation of artificial chromosome vectors includes bacterial artificial chromosomes (BACs), yeast artificial chromosomes (YACs), and mammalian artificial chromosomes (MACs).

A. Plasmid DNA as a Vector:
Plasmids are naturally occurring extra-chromosomal double-stranded circular DNA molecules that carry an origin of replication and replicate autonomously within bacterial cells. Almost all the bacteria have plasmids containing a low copy number (1-4 per cell) or a high copy number (10-100 per cell). The size of the plasmid varies from 1 to 500 kb. Usually plasmids contribute to about 0.5 to 5.0% of the total DNA of bacteria.
Plasmids are named with a system of uppercase letters and numbers, where the lowercase “p” stands for “plasmid.” The plasmid vector pBR322, constructed in 1974, was one of the first genetically engineered plasmids to be used in recombinant DNA. In the case of pBR322, the BR identifies the original constructors of the vector (Bolivar and Rodriquez), and 322 is the identification number of the specific plasmid. Some plasmids are given names of the places where they are discovered e.g. pUC is the plasmid from University of California.
pBR322- The most common Plasmid Vector:
pBR322 has a DNA sequence of 4,361 bp. It carries ampicillin resistant and tetracycline resistant genes as markers for identification. The plasmid has unique recognition sites for the action of restriction endonucleases, namely EcoRI, Hindlll, BamHI and Pstl. These early vectors were often of low copy number, meaning that they replicate to yield only one or two copies in each cell. pUC18, is a derivative of pBR322 (Fig. 10.2). This is a “high copy number” plasmid (500 copies per bacterial cell). Plasmid vectors are modified to contain a specific antibiotic resistance gene and a linker and adaptor.

B. Vectors for Cloning Large Pieces of DNA:
i. Bacteriophage Lambda (λ) as a Vector:
Bacteriophage lambda (λ) has been widely used in recombinant DNA since engineering of the first viral cloning vector in 1974. Phage λ vectors are particularly useful for preparing genomic libraries, because they can hold a larger piece of DNA than a plasmid vector. Today many variations of λ, vectors exist. Insertion vectors have unique restriction endonuclease sites that allow the cloning of small DNA fragments in addition to the phage λ. genome. These are often used for preparing cDNA expression libraries. Replacement vectors have paired cloning sites on either side of a central gene cluster. This central cluster contains genes for lysogeny and recombination, which are not essential for the lytic life cycle (Fig. 10.3).

The central gene cluster can be removed and foreign DNA inserted between the “arms.” All phage vectors used as cloning vectors have been disarmed for safety and can only function in special laboratory conditions. The recombinant viral particle infects bacterial host cells, in a process called “transduction”. The host cells lyse after phage reproduction, releasing progeny virus particles. The viral particles appear as a clear spot of lysed bacteria or “plaque” on an agar plate containing a lawn of bacteria. Each plaque represents progeny of a single recombinant phage and contains millions of recombinant phage particles. Most contemporary vectors carry a lacZ‘ gene allowing blue-white selection.
ii. Bacteriophage λ Vectors Permit Efficient Construction of Large DNA Libraries:
Vectors constructed from bacteriophage λ are about a thousand times more efficient than plasmid vectors in cloning large numbers of DNA fragments. For this reason, phage λ vectors have been widely used to generate DNA libraries, comprehensive collections of DNA fragments representing the genome or expressed mRNAs of an organism. Two factors account for the greater efficiency of phage λ as a cloning vector. Infection of E. coli host cells by λ virions occurs at about a thousand fold greater frequencies than transformation by plasmids. Many more λ clones transformed colonies can be grown and detected on a single culture plate. When a λ virion infects an E. coli cell, it can undergo a cycle of lytic growth during which the phage DNA is replicated and assembled into more than 100 complete progeny phage, which are released when the infected cell lyses.
A λ virion consists of a head, which contains the phage DNA genome, and a tail, which functions in infecting E. coli host cells. The λ genes encoding the head and tail proteins, as well as various proteins involved in phage DNA replication and cell lysis, are grouped in discrete regions of the ≈50-kb viral genome (Fig. 10.4). The central region of the λ genome, however, contains genes that are not essential for the lytic pathway. Removing this region and replacing it with a foreign DNA fragment up to ≈25 kb long yields a recombinant DNA that can be packaged in vitro to form phage capable of replicating and forming plaques on a lawn of E. coli host cells.
In vitro packaging of recombinant λ DNA, which mimics the in vivo assembly process, requires preassembled heads and tails as well as two viral proteins. It is technically feasible to use λ phage cloning vectors to generate a genomic library, that is, a collection of λ clones that collectively represent all the DNA sequences in the genome of a particular organism. However, such genomic libraries for higher eukaryotes present certain experimental difficulties.
First, the genes from such organisms usually contain extensive intron sequences and therefore are too large to be inserted intact into λ phage vectors. As a result, the sequences of individual genes are broken apart and carried in more than one λ clone (this is also true for plasmid clones). Moreover, the presence of introns and long inter- genic regions in genomic DNA often make it difficult to identify the important parts of a gene that actually encode protein sequences.
Thus for many studies, cellular mRNAs, which lack the noncoding regions present in genomic DNA, are a more useful starting material for generating a DNA library. In this approach, DNA copies of mRNAs, called complementary DNAs (cDNAs), are synthesized using reverse transcriptase and cloned in phage vectors. A large collection of the resulting cDNA clones, representing all the mRNAs expressed in a cell type is called a cDNA library.

C. Hybrid Plasmid-Phage Vectors:
Hybrid plasmid-phage vectors have been developed to overcome the inherent size limitation of the Ml3 cloning system and are now being widely used for applications such as DNA sequencing and the production of probes for use in hybridisation studies. One additional feature of phage vectors is that the technique of packaging in vitro is sequence independent, apart from the requirement of having the cos sites separated by DNA of packagable size (38- 51 kb). This has been exploited in the construction of special vectors that contain plasmid sequences joined to the cos sites of phage λ.
Such vectors are referred as cosmids (Fig. 10.5). They are relatively small (4-6 kb) and hence can accommodate cloned DNA fragments up to some 47 kb in size. As they lack phage genes, they behave as plasmids when introduced into E. coli by the packaging/infection mechanism of λ. Cosmid vectors therefore offer an apparently ideal system as a highly efficient and specific tool for introducing the recombinant DNA into the host cell, with a cloning capacity nearly two fold greater than the best λ replacement vectors.

However, they also suffer from disadvantages, as often the gains of using cosmids instead of phage vectors are offset by the losses in regard to the ease of use and further processing of cloned sequences. These vectors are essentially plasmids that contain the f1 (M13) phage origin of replication. When cells containing the plasmid are superinfected with phage, they produce single-stranded copies of the plasmid DNA and secrete them into the medium as M13-like particles. Vectors such as pEMBL9 or pBBR322 can accept DNA fragments of up to 10 kb.

D. Artificial Chromosome Vectors:
Bacterial artificial chromosomes (BACs) and yeast artificial chromosomes (YACs) are important tools for mapping and analysis of complex eukaryotic genomes. Much of the work on the Human Genome Project and other genome sequencing projects depends on the use of BACs and YACs, because they can hold greater than 300 kb of foreign DNA. BACs are constructed using the fertility factor plasmid (F factor) of E. coli as a starting point.
i. Bacterial Artificial Chromosome (BAC) Vectors:
Over the years, scientists have developed a number of different kinds of vectors for modifying the genetic constitution of bacteria. The bacterial artificial chromosome is one of the plasmid-based vectors. Plasmids are the extra chromosomal DNA. A scientist can alter a strain of bacteria using a BAC, and then compare the altered bacteria to an unaltered strain to discover what role the inserted genes play in cell biology.
Plasmids like the bacterial artificial chromosome are inserted into bacteria using a process called electroporation. Electroporation involves disturbing the cell membrane with an electric shock, which creates temporary openings through which molecules may be inserted. Forerunners to the BAC included modified plasmids namely cosmid and the fosmid.
In 1992 the first bacterial artificial chromosome was created by Hiroaki Shizuya, a researcher at the California Institute of Technology, by modifying a plasmid called F-factor. F-factor plasmids are used naturally by bacteria to transfer DNA from one cell to another during periods of environmental stress, in order to increase genetic variability and the likelihood of survival. Unlike its predecessors, the BAC could carry large genes with hundreds of thousands of DNA base pairs, or several genes at a time (Fig. 10.7).
ii. Yeast Artificial Ahromosome (YAC) Vectors:
Yeast is a single celled eukaryote that can be manipulated and grown in the lab much like bacteria. YAC vectors are designed to act like chromosomes. The yeast artificial chromosome (YAC) vector is capable of carrying a large DNA fragment (up to 2 Mb), however, its transformation efficiency is very low (Fig. 10.8).
A YAC can be considered as a functional artificial chromosome (self-replicating element), since it includes three specific DNA sequences that enable it to propagate from one cell to its offspring:
TEL:
The telomere which is located at each chromosome end protects the linear DNA from degradation by nucleases.
CEN:
The centromere which is the attachment site for mitotic spindle fibers “pulls” one copy of each duplicated chromosome into each new daughter cell.
ORI:
Replication origin sequences which are specific DNA sequences that allows the DNA replication machinery to assemble on the DNA and move at the replication forks.
It also contains few other specific sequences like selectable markers that allow the easy isolation of yeast cells that have taken up the artificial chromosome recognition site for the restriction enzymes.
 Vectors")
5. Essay on the Methods of Gene Transfer:
A. Transformation: Transfer of Recombinant Plasmid DNA to a Bacterial Host:
The traditional method of transformation is to incubate the cells in a concentrated calcium salt solution to make their membranes leaky. The permeable “competent” cells are then mixed with DNA to allow entry of the DNA into the bacterial cell. Successfully transformed bacteria carry either recombinant or non-recombinant plasmid DNA. Multiplication of the plasmid DNA occurs within each transformed bacterium. As the host cell divides, the plasmid vectors are passed on to progeny, where they continue to replicate. Numerous cell divisions of single transformed bacteria thus result in a clone of cells (visible as a bacterial colony) from a single parental cell. The cloned DNA can then be isolated from the clone of bacterial cells. Alternatively, a process called electroporation can be used that drives DNA into cells by a strong electric current.
B. Electroporation:
The uptake of free DNA can be induced by subjecting bacteria to a high voltage electric field in the presence of DNA. The experimental protocols for electroporation are different for various bacterial species. For E.coli, the cells (-50 µl) and DNA are placed in a chamber fitted with electrodes, and a single pulse of approximately 25 µF, 2.5 kv and 200 ohms is applied for about 4.6 milliseconds will yields transformation efficiencies of 109 transforments per microgram of DNA for small plasmids (-3 kb) and 106 for large plasmids (-136 kb). Similar conditions are used to introduce BAC vector DNA into E.coli.
The supposed mechanism of DNA uptake during electroporation is of chemically induced transformation, that transient pores are formed in the cell wall as a result of the electroshock and that, after contact with the lipid bilayer of the cell membrane, the DNA is taken into the cell.
6. Essay on the Recombinant Selection:
A. Selection by Marker Genes:
The selection of transformed cell depends on the particular vector, for example in the case of pUC18, a selectable marker gene for resistance to the antibiotic ampicillin. Culturing of cells in a medium containing ampicillin will allow only those cells containing the marker gene. The ampicillin resistance genes carried by the recombinant plasmids produce an enzyme, β-lactamase that cleaves a specific bond in the four-membered ring (β -lactam ring) in the ampicillin molecule that is essential to its antibiotic action (Table 10.5).

If the plasmid vector is introduced into a plasmid free antibiotic-sensitive bacterial cell, the cell becomes resistant to ampicillin. Non-transformed cells contain no pUC18 DNA, therefore they will not be antibiotic-resistant, and their growth will be inhibited on agar containing ampicillin. Transformed bacterial cells may contain either non-recombinant pUC18 DNA (self-ligated vector only) or recombinant pUC18 DNA (vector containing foreign DNA insert). Both types of transformed bacterial cells will be ampicillin-resistant (Fig. 10.10).

B. Blue-White Screening:
To distinguish nonrecombinant from recombinant transformants, blue-white screening or “lac selection” (also called α-complementation) can be used with vector containing lacZ gene. Bacterial colonies are grown on selective medium containing ampicillin and a colorless chromogenic compound called X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside). pUC 18 carries a portion of the lacZ gene (called lacZ ‘) that encodes the first 146 amino acids for the enzyme β-galactosidase nonfunctional proteins.
The multiple cloning sites reside in the coding region. If the lacZ.’region is not interrupted by inserted DNA, the ami- noterminal portion of β-galactosidase is synthesized. However, by a-complementation the two partial proteins can associate and form a functional enzyme. When present the enzyme p -galactosidase, catalyzes hydrolysis of X-gal, converting the colorless substrate into a blue-colored product (Fig. 10.11).
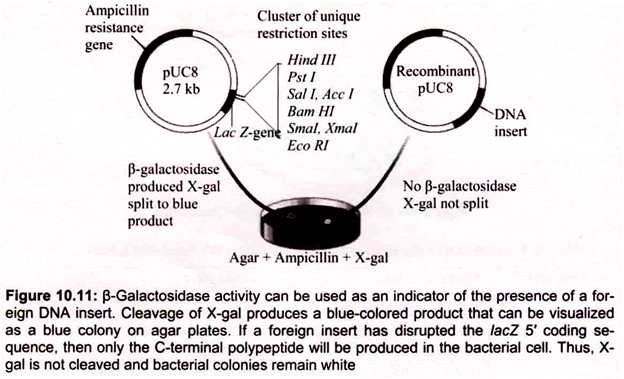
Further screening of positive (white) colonies can be done by restriction endonuclease digest to confirm the presence and orientation of the insert. When a positive colony containing recombinant plasmid DNA is transferred aseptically to liquid growth medium, the cells continue to multiply exponentially. Within a day or two, a culture containing trillions of identical cells can be harvested.
The final step in molecular cloning is the recovery of the cloned DNA. Plasmid DNA can be purified from crude cell lysates by chromatography using silica gel or anion exchange resins that preferentially bind nucleic acids under appropriate conditions and allow for the removal of proteins and polysaccharides. The purified plasmid DNA can then be eluted and recovered by ethanol precipitation in the presence of monovalent cations. Ethanol precipitation of plasmid DNA from aqueous solutions yields a clear pellet that can be easily dissolved in an appropriate buffered solution.
7. Essay on the Expression Systems:
Prokaryotic (bacteria) or eukaryotic (yeast, mammalian cell culture) systems are generally used as a host for the production of usable quantities of the desired rDNA product. Most of the rDNA products approved by FDA are being produced using these systems. Bacteria such as Escherichia coli are widely used for the expression of rDNA products. They offer several advantages due to high level of recombinant protein expression, rapid growth of cell and simple media requirement.
However, there are some limitations such as intracellular accumulation of heterologous proteins, the potential for product degradation due to trace of protease impurities and production of endotoxin. Yeast such as Saccharomyces cerevisiae, Hansenulla polymorpha and Pichia pastoris are among the simplest eukaryotic organisms. They grow relatively quickly and are highly adaptable to large-scale production. These organisms do not produce endotoxin. They are capable of glycosylating proteins up to a certain extent like mammalian cells.
Mammalian systems such as Chinese hamster ovary (CHO) cell and baby hamster kidney (BHK) cell systems are often the choice for production of human therapeutic proteins. The CHO and BHK cell systems are an ideal choice as these are capable of glycosylating the protein at the correct sites. However, cost of production of the products using these systems is high because of slow growth and expensive nutrient media. The choice of expression system can influence the character, quantity and cost of a final product.
Recent advances have been made in producing therapeutic proteins by using transgenic animals. Transgenic milk production is currently most feasible. The advantages of this system are high expression levels and volume output, low capital investment, low operational costs and reproducible production facility, i.e. inbreeding could pass an animal’s ability to produce transgenic protein to its offspring. Despite the attractiveness of this system, a number of issues remain to be solved before it is broadly accepted by the industries and regulatory authorities alike.
These include variability of expression levels and characterization of the exact nature of the post-translational modification in the mammary systems. The use of genetically engineered plants to produce valuable proteins is increasing slowly. The system has potential advantages of economy and scalability. However, variation in product yield, contamination with agrochemical and fertilizer, impact of pest and disease, and variable cultivation conditions should also be considered. Plant cell culture system combines the advantages of whole plant system as well as animal cell culture. Although, no recombinant products have yet been produced commercially using plant cell culture, several companies are investigating the commercial feasibility of such a production system.
8. Essay on the Production and Purification of rDNA Products:
Recombinant DNA (rDNA) technology has made a revolutionary impact in the area of human healthcare by enabling mass production of safe, pure and effective rDNA expression products. As we have seen for the production of a desired rDNA product the gene of interest has to be identified and isolated. After isolation and characterization of the gene, it is inserted into a vector such as plasmid.
The recombinant plasmid is then inserted into bacterial yeast or cultured animal cell. Clones of transformed host cell are isolated and those that produce the protein of interest in the desired quantities are preserved under suitable condition as a master cell bank. The cell banks are characterized and properly maintained for use in subsequent transformation procedures.
The cell bank should be periodically tested for cell viability, genetic and phenotypic stability. When eukaryotic cells are used for production, distinguishing genetic, phenotypic and immunological markers of the cell are useful in establishing the identity of the cells. Likewise, when microbial cultures are used, specific phenotypic features which form a basis for identification should be described.
As manufacturing needs arise, cells from working cell can be scaled up to produce the product in roller bottles and/or fermentors. Traditional small-scale fermentors are generally used for expansion of cells in suspension culture such as the growth of E. coli or yeast. The presence, extent and nature of any microbial contamination in the culture vessels must be thoroughly examined at suitable stages during production. All the systems associated with fermentors must be validated before being routinely used.
Mammalian cells are often grown in roller bottles. Inoculation of host cells that contain an expression vector is added to defined volume of medium in either fermentors or roller bottles. The cells are allowed to grow until the nutrients in the medium are depleted or excreted by the products reaching inhibitory levels. By providing a balanced mixture of nutrients and/or chemicals to neutralize accumulating growth inhibitor, product yield in the medium or cell density can be improved. At the end of the run, the host cells are harvested and the recombinant product is isolated from culture medium or cells.
One of the main problems faced in industrial production of rDNA products is plasmid instability in continuous and large-scale fermentation, since these cultures go through many generations. The resulting effects are lower productivity and an increase in production cost, because of the build-up of non-productive plasmid free cells. Unintended variability in culture during production may lead to changes which cause alteration in the product. Such variations might result in differing yield of the products. Thus the steps in a production process should be validated to ensure that the process intermediates are within specifications. Any assay used during process validation must itself be standardized, before the process validation is commenced.
High level expression of rDNA products in different host systems can often result in aggregation and accumulation of inclusion bodies. Under appropriate conditions, the rDNA products may get deposited in inclusion bodies, approximately 50% or more of the total cell protein. The rDNA protein is highly pure, stable and compact and can be recovered from the inclusion bodies by proper techniques. The first step is the isolation of the inclusion body from disrupted cells by centrifugation.
The inclusion body proteins were then solubilized by using a chaotrope (denaturant), a substance which disrupts the three dimensional structure in macromolecules such as proteins, DNA, or RNA and denatures them. Chaotropic agents such as urea and guanidinium chloride interfere with stabilizing inter-molecular interactions mediated by non-covalent forces such as hydrogen bonds, van der waals forces, and hydrophobic effects. The active protein will be then recovered by removal of denaturant under controlled conditions that allow the protein to adapt its native configuration.
The overall goal of purification is to bring as much product with as little loss as possible. In case the desired product is intracellular, fermentation is followed by harvesting of cells. This step is normally achieved by centrifugation or filtration. The cells are then disrupted or lysed and cell debris is removed by centrifugation, leaving behind a dilute solution of crude desired product.
If mammalian or yeast system is used, the desired product can be obtained directly from the medium. Nowadays, ultrafiltration has become the method of choice for concentration and chromatographic techniques for purification of rDNA products. Among the different chromatographic techniques generally employed in a typical downstream process, gel filtration and ion exchange chromatography are the most common. Affinity chromatography is employed wherever possible, as it has high bio-specificity and one can achieve a high degree of purification.
Physico-chemical, biological and immunological characterizations of purified product should be ensured using a wide range of analytical tests. The purification process must be validated to ensure that it is effective and adequate to remove extraneous substances such as chemicals used in purification, column contaminations, endotoxin, residual cellular proteins and viruses. Analytical methods play a vital role in the determination of identity, purity and potency of rDNA products with respect to safe and efficacious medicine for human use. Characterization of products may include test for mainly amino acid composition, peptide mapping, electrophoretic assays (SDS-PAGE, Western blot, isoelectric focusing), HPLC, immunoassay, endotoxin test, potency determination test, etc.
Bicinchoninic acid and Bradford methods are generally used for the determination of protein concentration in rDNA product. Protein sequencing provides structure information. Amino acid analysis is used to identify rDNA products based on their amino acid composition. Peptide mapping is used to compare protein structure of the product with that of the reference standard and to confirm lot-to-lot consistency of primary structure.
Electrophoretic assays (sodium dodecylsulphate polyacrylamide gel electrophoresis, Western blot and isoelectric focusing) are effectively used to evaluate rDNA product purity, identity, homogeneity and stability. More recently, capillary electrophoresis has generated considerable interest as a complementary technique in the analysis of rDNA products.
Chromatographic method, viz. reverse phase-HPLC, size exclusion chromatography and hydrophobic interaction chromatography are used in the determination of purity of the product as well as the level of known impurities or degradation of product. Immunoassay and DNA hybridization are used for determination of host cell impurities. Polymerase chain reaction, which involves DNA amplification, may prove useful in detection and identification of contaminant DNA. The limulus amebocyte lysate (LAL) test is the most sensitive and specific test used to detect and measure endotoxin. A comparative study demonstrated that LAL test is more sensitive than rabbit pyrogen test.
The potency of rDNA product is generally assessed by using specific techniques. The choice of assay is often determined by the nature of the product and its intended therapeutic application. Animal model assay, cell culture-based assay and in vitro (physico-chemical) assays used for determination of potency of rDNA product involves comparing the activity of the recovered product with that of reference standard (calibrated in international unit) and interpretation of the data with statistical support.
Potency assays should have wide confidence limits and may include animal-based bioassay, in vitro cell culture assays or biochemical assay. All rDNA drugs undergo rigorous quality control testing in order to conform to predetermined specifications. The fundamental difference between quality control system for rDNA drugs and traditional pharmaceuticals lies in the types of method used to determine drug identity, purity and potency. The use of suitable reference standard from internationally recognized sources such as WHO, NIH, etc. is important for identification and purity of rDNA-derived drugs.
9. Essay on Transgenic Plants:
For production of transgenic animals, DNA is usually microinjected into pronuclei of embryonic cells at a very early stage after fertilization, or alternatively gene targeting of embryo stem (ES) cells is employed. This is possible in animals due to the availability of specialized in vitro fertilization technology, which allows manipulation of ovule, zygote or early embryo. Such techniques are not available in plants. In contrast to this in higher plants, cells or protoplasts can be cultured and used for regeneration of whole plants.
Therefore, these protoplasts can be used for gene transfer followed by regeneration leading to the production of transgenic plants. Besides cultured cells and protoplasts, other meristem cells (immature embryos or organs), pollen or zygotes can also be used for gene transfer in plants. To achieve genetic transformation in plants, we need the construction of a vector (genetic vehicle) which transports the genes of interest, flanked by the necessary controlling sequences i.e. promoter and terminator, and deliver the genes into the host plant.
A. Methods of Gene Transfer in Plants:
The two kinds of gene transfer methods in plants are:
i. Vector-Mediated or Indirect Gene Transfer:
Among the various vectors used in plant transformation, the Ti plasmid of Agrobacterium tumefaciens has been widely used. This bacteria is known as “natural genetic engineer” of plants because these bacteria have natural ability to transfer T-DNA of their plasmids into plant genome upon infection of cells at the wound site and cause an unorganized growth of a cell mass known as crown gall. Ti plasmids are used as gene vectors for delivering useful foreign genes into target plant cells and tissues (Fig. 10.12). The foreign gene is cloned in the T-DNA region of Ti-plasmid in place of unwanted sequences.

To transform plants, leaf discs (in case of dicots) or embryogenic callus (in case of monocots) are collected and infected with Agrobacterium carrying recombinant disarmed Ti- plasmid vector. The infected tissue is then cultured (co-cultivation) on shoot regeneration medium for 2-3 days during which time the transfer of T-DNA along with foreign genes takes place.
After this, the transformed tissues (leaf discs/calli) are transferred onto selection cum plant regeneration medium supplemented with usually lethal concentration of an antibiotic to selectively eliminate non-transformed tissues. After 3-5 weeks, the regenerated shoots (from leaf discs) are transferred to root-inducing medium, and after another 3-4 weeks, the complete plants are transferred to soil following the hardening (acclimatization) of regenerated plants. The molecular techniques like PCR and southern hybridization are used to detect the presence of foreign genes in the transgenic plants.
ii. Vector- Less or Direct Gene Transfer:
In this method, the foreign gene of interest is delivered into the host plant cell without the help of a vector.
The methods used for direct gene transfer in plants are:
a. Chemical mediated gene transfer e.g. chemicals like polyethylene glycol (PEG) and dextran sulphate induce DNA uptake into plant protoplasts. Calcium phosphate is also used to transfer DNA into cultured cells.
b. Microinjection, where the DNA is directly injected into plant protoplasts or cell (specifically into the nucleus or cytoplasm) using fine tipped (0.5 – 1.0 µm diameter) glassneedle or micropipette. This method of gene transfer is used to introduce DNA into large cells such as oocytes, eggs, and the cells of early embryo.
c. Electroporation, involves a pulse of high voltage applied to protoplasts/cells/ tissues to make transient (temporary) pores in the plasma membrane which facilitates the uptake of foreign DNA. The cells are placed in a solution containing DNA and subjected to electrical shocks to cause holes in the membranes. The foreign DNA fragments enter through the holes into the cytoplasm and then to nucleus.
d. Particle gun/Particle bombardment – In this method, the foreign DNA containing the genes to be transferred is coated onto the surface of minute gold or tungsten particles (1- 3 µm) and bombarded onto the target tissue or cells using a particle gun (also called as gene gun/shot gun/microprojectile gun). The microprojectile bombardment method was initially named as biolistics by its inventor, Sanford (1988). Two types of plant tissue are commonly used for particle bombardment- Primary explants and the proliferating embryonic tissues.
e. Transformation – This method is used for introducing foreign DNA into bacterial cells e.g. E. coli. The transformation frequency (the fraction of cell population that can be transferred) is very good in this method, e.g. the uptake of plasmid DNA by E. coli is carried out in ice cold CaCl2 (0-5°C) followed by heat shock treatment at 37-45°C for about 90 sec. The transformation efficiency refers to the number of transformants per microgram of added DNA. The CaCl2 breaks the cell wall at certain regions and binds the DNA to the cell surface.
f. Liposome mediated gene transfer or Lipofection – Liposomes are circular lipid molecules with an aqueous interior that can carry nucleic acids. Liposomes encapsulate the DNA fragments and then adher to the cell membranes and fuse with them to transfer DNA fragments. Thus, the DNA enters the cell and then to the nucleus. Lipofection is very efficient technique used to transfer genes in bacterial, animal and plant cells.
B. Beneficial Traits of Transgenic Plants:
During the last decades, a tremendous progress has been made in the development of transgenic plants using the various techniques of genetic engineering. The plants, in which a functional foreign gene has been incorporated by any biotechnological methods that generally not present in the plant, are called transgenic plants. Transgenic plants have many beneficial traits like insect resistance, herbicide tolerance, delayed fruit ripening, improved oil quality, weed control etc.
Some of the traits introduced in these transgenic plants are as follows:
i. Stress Tolerance:
Biotechnology strategies are being developed to overcome problems caused due to biotic stresses (viral, bacterial infections, pests and weeds) and abiotic stresses (physical factors such as temperature, humidity, salinity etc.).
ii. Abiotic Stress Tolerance:
The plants show their abiotic stress response reactions by the production of stress related osmolytes like sugars (e.g. trehalose and fructans), sugar alcohols (e.g. mannitol), amino acids (e.g. proline, glycine, betaine) and certain proteins (e.g. antifreeze proteins). Transgenic plants have been produced which over express the genes for one or more of the above mentioned compounds. Such plants show increased tolerance to environmental stresses. Resistance to abiotic stresses includes stress induced by herbicides, temperature (heat, chilling, freezing), drought, salinity, ozone and intense light. These environmental stresses result in the destruction, deterioration of crop plants which leads to low crop productivity. Several strategies have been used and developed to build resistance in the plants against these stresses.
iii. Herbicide Tolerance:
Several biotechnological strategies for weed control are being used e.g. the over-production of herbicide target enzyme (usually in the chloroplast) in the plant which makes the plant insensitive to the herbicide. This is done by the introduction of a modified gene that encodes for a resistant form of the enzyme targeted by the herbicide in weeds and crop plants.
The biological manipulations using genetic engineering to develop herbicide resistant plants are:
(a) Over-expression of the target protein by integrating multiple copies of the gene or by using a strong promoter,
(b) Enhancing the plant detoxification system which helps in reducing the effect of herbicide,
(c) Detoxifying the herbicide by using a foreign gene, and
(d) Modification of the target protein by mutation.
Some of the examples are:
(i) Glyphosate Resistance:
Glyphosate, a glycine derivative is a herbicide which is found to be effective against the 76 of the world’s worst 78 weeds. It kills the plant by being the competitive inhibitor of the enzyme 5-enoyl-pyruvylshikimate 3- phosphate synthase (EPSPS) in the shikimic acid pathway. Due to its structural similarity with the substrate phosphoenol pyruvate, glyphosate binds more tightly with EPSPS and thus blocks the shikimic acid pathway. Certain strategies were used to provide glyphosate resistance to plants. It was found that EPSPS gene was over-expressed in Petunia due to gene amplification. EPSPS gene was isolated from Petunia and introduced in to the other plants. These plants could tolerate glyphosate at a dose of 2-4 times higher than that required to kill wild type plants.
(ii) Phosphinothricin is a broad spectrum herbicide and is effective against broad-leafed weeds. It acts as a competitive inhibitor of the enzyme glutamine synthase which results in the inhibition of the enzyme glutamine synthase and accumulation of ammonia and finally the death of the plant. The disturbace in the glutamine synthesis also inhibits the photosynthetic activity. The enzyme phosphinothricin acetyl transferase (which was first observed in Streptomyces sp in natural detoxifying mechanism against phosphinothricin) acetylates phosphinothricin, and thus inactivates the herbicide. The gene encoding for phosphinothricin acetyl transferase (bar gene) was introduced in transgenic maize and oil seed rape to provide resistance against phosphinothricin.
iv. Other Abiotic Stresses:
The abiotic stresses due to temperature, drought, and salinity are collectively also known as water deficit stresses. The plants produce osmolytes or osmoprotectants to overcome the osmotic stress. The attempts are on to use genetic engineering strategies to increase the production of osmoprotectants in the plants. The biosynthetic pathways for the production of many osmoprotectants have been established and genes coding the key enzymes have been isolated e.g. Glycine betaine is a cellular osmolyte which is produced by the participation of a number of key enzymes like choline dehydrogenase, choline monooxygenase etc.
The choline oxidase gene from Arthrobacter sp. was used to produce transgenic rice with high levels of glycine betaine giving tolerance against water deficit stress. Scientists also developed cold- tolerant genes (around 20) in Arabidopsis when this plant was gradually exposed to slowly declining temperature. By introducing the coordinating gene (it encodes a protein which acts as transcription factor for regulating the expression of cold tolerant genes), expression of cold tolerant genes was triggered giving protection to the plants against the cold temperatures.
(i) Insect Resistance:
A variety of insects, mites and nematodes significantly reduce the yield and quality of the crop plants. The conventional method is to use synthetic pesticides, which also have severe effects on human health and environment. The transgenic technology uses an innovative and eco-friendly method to improve pest control management. About 40 genes obtained from microorganisms of higher plants and animals have been used to provide insect resistance in crop plants. The first genes available for genetic engineering of crop plants for pest resistance were Cry genes (popularly known as Bt genes) obtained from a bacterium Bacillus thuringiensis.
These are specific to particular group of insect pests and are not harmful to other useful insects like butter flies and silk worms. Transgenic crops with Bt genes (e.g. cotton, rice, maize, potato, tomato, brinjal, cauliflower, cabbage, etc.) have been developed. This has proved to be an effective way of controlling the insect pests and has reduced the pesticide use.
The most notable example is Bt cotton (which contains CrylAc gene) that is resistant to a notorious insect pest Bollworm (Helicoperpa armigera). Certain genes from higher plants were also found to result in the synthesis of products possessing insecticidal activity. One of the examples is the Cowpea trypsin inhibitor gene (CpTi) which was introduced into tobacco, potato, and oilseed rape for develping transgenic plants. Cowpea trypsin inhibitor (CpTi) has no effect on mammalian trypsin, hence it is non-toxic to mammals.
(ii) Virus Resistance:
There are several strategies for engineering plants for viral resistance, and these utilizes the genes from virus itself (e.g. the viral coat protein gene). The virus-derived resistance has given promising results in a number of crop plants such as tobacco, tomato, potato, alfalfa, and papaya. The induction of virus resistance is done by employing virus-encoded genes- virus coat proteins, movement proteins, transmission proteins, satellite RNA, antisense RNAs, and ribozymes.
The virus coat protein-mediated approach is the most successful one to provide virus resistance to plants. It was in 1986, transgenic tobacco plants expressing tobacco mosaic virus (TMV) coat protein gene were first developed. These plants exhibited high levels of resistance to TMV. The transgenic plant providing coat protein-mediated resistance to virus is rice, potato, peanut, sugar beet, alfalfa, etc.
(iii) Resistance against Fungal and Bacterial Infections:
As a defence strategy against the invading pathogens (fungi and bacteria) the plants accumulate low molecular weight proteins which are collectively known as pathogenesis-related (PR) proteins. Several transgenic crop plants with increased resistance to fungal pathogens are being raised with genes coding for the different compounds. One of the examples is the Glucanase enzyme that degrades the cell wall of many fungi.
The most widely used glucanase is β-1,4-glucanase. The gene encoding for β-1,4 glucanase has been isolated from barley, introduced, and expressed in transgenic tobacco plants. This gene provided good protection against soil-borne fungal pathogen Rhizoctonia solani. Lysozyme degrades chitin and peptidoglycan of cell wall and in this way fungal infection can be reduced. Transgenic potato plants with lysozyme gene providing resistance to Erwinia carotovora have been developed.
v. Delayed Fruit Ripening:
The ripening can be slowed down by blocking or reducing ethylene production since ethylene regulates the ripening of fruits. This can be achieved by introducing ethylene forming gene(s) in a way that will suppress its own expression in the crop plant. Such fruits ripen very slowly (however, they can be ripen by ethylene application) and this helps in exporting the fruits to longer distances without spoilage due to longer-shelf life. The main strategy used was the antisense RNA approach.
In the normal tomato plant, the PG gene (for the enzyme polygalacturonase) encodes a normal mRNA that produces the enzyme polygalacturonase which is involved in the fruit ripening. The complimentary DNA of PG encodes for antisense mRNA, which is complimentary to normal (sense) mRNA. The hybridization between the sense and antisnse mRNAs renders the sense mRNA ineffective. Consequently, polygalacturonase is not produced causing delay in the fruit ripening. Similarly strategies have been developed to block the ethylene biosynthesis thereby reducing the fruit ripening.
vi. Male Sterility:
The plants may inherit male sterility either from the nucleus or cytoplasm. It is possible to introduce male sterility through genetic manipulations while the female plants maintain fertility. In tobacco plants, these are created by introducing a gene coding for an enzyme (barnase, which is a RNA hydrolyzing enzyme) that inhibits pollen formation. This gene is expressed specifically in the tapetal cells of anther using tapetal specific promoter TA29 to restrict its activity only to the cells involved in pollen production. The restoration of male fertility is done by introducing another gene barstar that suppresses the activity of barnase at the onset of the breeding season. By using this approach, transgenic plants of tobacco, cauliflower, cotton, tomato, corn etc. with male sterility have been developed.
10. Essay on the Edible Vaccines:
Vaccines have been revolutionary for the prevention of infectious diseases. Edible vaccines hold great promise as a cost-effective, easy-to-administer, easy-to-store, fail-safe and socio- culturally readily acceptable vaccine delivery system, especially for the poor developing countries. It involves introduction of selected desired genes into plants and then inducing these altered plants to manufacture the encoded proteins.
Crop plants offer cost-effective bioreactors to express antigens which can be used as edible vaccines. The approach is to isolate genes encoding antigenic proteins from the pathogens and then expressing them in plants. Such transgenic plants or their tissues producing antigens can be eaten for vaccination/immunization (edible vaccines). The expression of such antigenic proteins in crops like banana and tomato are useful for immunization of humans since banana and tomato fruits can be eaten raw.
Transgenic plants (tomato, potato) have been developed for expressing antigens derived from animal viruses e.g. rabies virus and herpes virus. In 1990, the first report of the production of edible vaccine (a surface protein from Streptococcus) in tobacco at 0.02% of total leaf protein level was published in the form of a patent application under the International Patent Cooperation Treat.The first clinical trials in humans, using a plant derived vaccine were conducted in 1997 and were met with limited success. This involved the ingestion of transgenic potatoes with a toxin of E. coli causing diarrhea.
Creating edible vaccines involves introduction of selected desired genes into plants and then inducing these altered plants to manufacture the encoded proteins. This process is known as “transformation”, and the altered plants are called “transgenic plants”. The process of making of edible vaccines involves the incorporation of a plasmid carrying the antigen gene and an antibiotic resistance gene, into the bacterial cells, e.g. Agrobacterium tumefaciens.
The small pieces of potato leaves are exposed to an antibiotic which can kill the cells that lack the new genes. The surviving cells with altered genes multiply and form a callus. This callus is allowed to grow and subsequently transferred to soil to form a complete plant. In about a few weeks, the plants bear potatoes with antigen vaccines.
Like conventional subunit vaccines, edible vaccines are composed of antigenic proteins and are devoid of pathogenic genes. Thus, they have no way of establishing infection, assuring its safety, especially in immunocompromised patients. The antigens in transgenic plants are delivered through bio-encapsulation, i.e., the tough outer wall of plant cells, which protects them from gastric secretions and finally break up in the intestines. The antigens are released, taken up by M cells in the intestinal lining that overlie Peyer’s patches and gut- associated lymphoid tissue (GALT), passed on to macrophages, other antigen-presenting cells; and local lymphocyte populations, generating serum IgG, IgE responses, local IgA response and memory cells, which would promptly neutralize the attack by the real infectious agent.
Conventional subunit vaccines are expensive and technology-intensive, need purification, require refrigeration and produce poor mucosal response. In contrast, edible vaccines would enhance compliance, especially in children and because of oral administration, would eliminate the need for trained medical personnel. Their production is highly efficient and can be easily scaled up. The edible vaccines produced in transgenic plants will solve the storage problems, will ensure easy delivery system by feeding and will have low cost as compared to the recombinant vaccines produced by bacterial fermentation.
The bacteria E. coli, V. cholerae cause acute watery diarrhea by colonizing the small intestine and by producing toxins. Another strategy adopted to produce a plant-based vaccine, is to infect the plants with recombinant virus carrying the desired antigen that is fused to viral coat protein. The infected plants are reported to produce the desired fusion protein in large amounts in a short duration. The technique involves either placing the gene downstream a subgenomic promoter, or fusing the gene with capsid protein that coats the virus.
11. Essay on Gene Therapy:
Genes are specific sequences of bases that encode instructions on how to make proteins. Although genes get a lot of attention, it’s the proteins that perform most life functions and even make up the majority of cellular structures. When genes are altered so that the encoded proteins are unable to carry out their normal functions, genetic disorders can result. Gene therapy is a technique for correcting defective genes responsible for disease development.
Researchers may use one of several approaches for correcting faulty genes such as a normal gene may be inserted into a non-specific location within the genome to replace a nonfunctional gene. This approach is most common. An abnormal gene could be swapped for a normal gene through homologous recombination. The abnormal gene could be repaired through selective reverse mutation, which returns the gene to its normal function. The regulation (the degree to which a gene is turned on or off) of a particular gene could be altered.
In most gene therapy studies, a “normal” gene is inserted into the genome to replace an “abnormal,” disease-causing gene. A carrier molecule called a vector must be used to deliver the therapeutic gene to the patient’s target cells. Currently, the most common vector is a virus that has been genetically altered to carry normal human DNA. Viruses have evolved a way of encapsulating and delivering their genes to human cells in a pathogenic manner.
Scientists have tried to take advantage of this capability and manipulate the virus genome to remove disease-causing genes (virulent) and insert therapeutic genes. Target cells such as the patient’s liver or lung cells are infected with the viral vector. The vector then unloads its genetic material containing the therapeutic human gene into the target cell. The generation of a functional protein product from the therapeutic gene restores the target cell to a normal state.
Some of the different types of viruses used as gene therapy vectors are:
A. Retroviruses:
They are the classes of viruses like Human immunodeficiency virus (HIV) that can create double-stranded DNA copies of their RNA genomes. These copies of its genome can be integrated into the chromosomes of host cells.
B. Adenoviruses:
A class of viruses with double-stranded DNA genomes that cause respiratory, intestinal, and eye infections in humans.
C. Adeno-Associated Viruses:
A class of small, single-stranded DNA viruses that can insert their genetic material at a specific site on chromosome 19.
D. Herpes Simplex Viruses:
A class of double-stranded DNA viruses that infect a particular cell type, neurons. Herpes simplex virus type 1 is a common human pathogen that causes cold sores.
Besides virus-mediated gene-delivery systems, there are several non-viral options for gene delivery. The simplest method is the direct introduction of therapeutic DNA into target cells. This approach is limited in its application because it can be used only with certain tissues and requires large amounts of DNA. It consists of approaches which involve the creation of an artificial lipid sphere with an aqueous core.
This liposome, which carries the therapeutic DNA, is capable of passing the DNA through the target cell’s membrane. Therapeutic DNA also can get inside target cells by chemically linking the DNA to a molecule that will bind to special cell receptors. Once bound to these receptors, the therapeutic DNA constructs are engulfed by the cell membrane and passed into the interior of the target cell. This delivery system tends to be less effective than other options.
The Food and Drug Administration (FDA) has not yet approved any human gene therapy product for sale. Current gene therapy is experimental and has not proven very successful in clinical trials. Little progress has been made since the first gene therapy clinical trial began in 1990. In 1999, gene therapy suffered a major setback with the death of 18-year-old Jesse Gelsinger. Jesse was participating in a gene therapy trial for ornithine transcarboxylase deficiency (OTCD). He died from multiple organ failures 4 days after starting the treatment. His death is believed to have been triggered by a severe immune response to the adenovirus carrier.